- Riconoscere i segni tardivi (linguaggio, motricità, deglutizione) è essenziale per sicurezza e assistenza.
- Diagnosi supportata da test cognitivi, imaging PET e biomarcatori di Aβ e tau.
- Terapie sintomatiche (AChEI, memantina) e interventi non farmacologici con benefici moderati.
- Fattori vascolari e stile di vita influenzano il decorso; genetica e biomarcatori guidano il futuro.
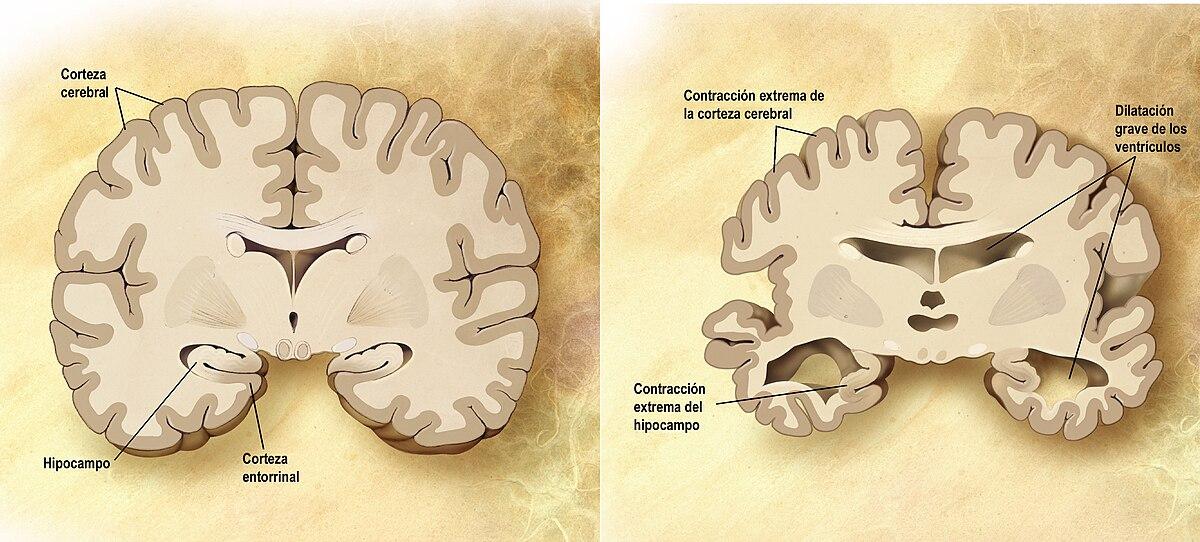
L’Alzheimer è una malattia neurodegenerativa progressiva che, nel tempo, compromette memoria, linguaggio, giudizio, capacità di risolvere problemi, personalità e movimento. Non è un semplice “invecchiamento normale”: si tratta di un processo patologico che si sviluppa nell’arco di anni e colpisce aree cerebrali sempre più estese, con ripercussioni significative sulla vita quotidiana della persona e della famiglia.
Comprendere le fasi e i segni, in particolare quelli tardivi, è cruciale per riconoscere i cambiamenti, pianificare l’assistenza e ridurre i rischi (cadute, malnutrizione, infezioni). Le “fasi” sono linee guida utili, ma ogni storia clinica è unica: l’evoluzione varia da individuo a individuo, e i sintomi possono sovrapporsi o progredire a velocità differenti.
Che cos’è l’Alzheimer e come progredisce
La progressione è molto variabile: in media, dopo la diagnosi si vive tra 3 e 11 anni, ma sono possibili durate oltre i 20 anni. Lo stadio alla diagnosi e la presenza di fattori vascolari non controllati (per esempio ipertensione) possono accelerare l’andamento clinico. Tra le cause comuni di morte nelle fasi avanzate figurano la polmonite (spesso da aspirazione), la disidratazione, la malnutrizione, le cadute e altre infezioni.
Le fasi: dalla preclinica alla demenza grave
Fase preclinica (presintomatica). Inizia molto prima dei sintomi evidenti. Non si notano cambiamenti nel quotidiano, e anche i familiari non percepiscono alterazioni; tuttavia, biomarcatori e tecniche di imaging possono rilevare precocemente placche di beta-amiloide e grovigli di tau. In questa fase, che può durare anni o decenni, si stanno studiando trattamenti potenziali per ritardare la comparsa dei sintomi.
Lieve deterioramento cognitivo (MCI) attribuibile all’Alzheimer. Si osservano piccoli cambiamenti di memoria e attenzione che non compromettono ancora in modo marcato lavoro e relazioni. Possono emergere difficoltà nel ricordare informazioni appena apprese, organizzare i passi di un compito o stimare tempi e quantità; non tutti i MCI evolvono in Alzheimer, e i biomarcatori aiutano a chiarire l’eziologia.
Demenza lieve. In genere qui arriva la diagnosi. I problemi di memoria e pensiero iniziano a impattare le attività quotidiane in modo evidente per medici e familiari. Si intensificano domande ripetute, errori nella gestione di denaro e cali di giudizio, con cambiamenti di personalità (più apatia, irritabilità) e difficoltà a trovare parole chiare per esprimersi.
- Dimenticanza di eventi recenti e ripetizioni delle stesse domande.
- Problemi nel pianificare/risolvere compiti complessi (per es. bilanci familiari).
- Variazioni caratteriali (chiusura, irritabilità, scarsa motivazione).
- Difficoltà a nominare oggetti e a spiegare idee in modo coerente.
- Smarriti frequenti di oggetti e disorientamento anche in luoghi noti.
Demenza moderata. Crescono confusione e smarrimento; la persona può perdere l’orientamento su luogo, tempo e persone, arrivando a scambiare conoscenti o a non riconoscere contesti familiari. Aumenta il bisogno di aiuto nelle attività di cura personale, e possono comparire idee di riferimento sospettose, allucinazioni, agitazione serale e, talvolta, comportamenti fisicamente aggressivi.
- Giudizio compromesso e confusione profonda, con tendenza al vagabondaggio (pericoloso restare soli).
- Dimenticanze biografiche (indirizzo, numero di telefono, scuole frequentate) e confabulazioni per colmare i vuoti.
- Assistenza necessaria per vestirsi in base a clima/contesto, igiene, bagno e altre ADL; possibili incontinenze.
- Disturbi comportamentali: sospettosità, accuse infondate, allucinazioni, agitazione, soprattutto a fine giornata.
Demenza grave. Si perde la capacità di comunicare in modo comprensibile, con necessità di assistenza totale per tutte le attività di base. Le abilità motorie declinano: non si cammina più senza aiuto, talvolta non si riesce a restare seduti o a tenere il capo eretto; possono comparire rigidità muscolare e riflessi alterati. Con il tempo si perde la capacità di deglutire e il controllo vescica/intestino.
Segni precoci e come distinguerli dalla normale età
Non tutti gli “sbagli di memoria” sono patologici. Dimenticare un nome ma ricordare chi sia quella persona rientra nell’età; nell’Alzheimer, invece, sfuma anche il contesto: non solo non si ricorda il nome, ma spesso non si colloca la relazione (non riconoscere un vicino o un commerciante).
Memoria e concentrazione: cambio di posto o perdita di oggetti importanti, confusione su compiti quotidiani, difficoltà con conti semplici e decisioni routinarie, disorientamento su mese o stagione, problemi con il test dell’orologio, la costruzione tridimensionale e l’orientamento in spazi aperti.
Umore e condotta: sbalzi d’umore imprevedibili, perdita d’interesse per hobby e ambiente, depressione/ansia o confusione ai cambiamenti, negazione dei sintomi, possibili irritabilità, agitazione, disinibizione sociale.
Segni tardivi dell’Alzheimer
Linguaggio e discorso: difficoltà a completare frasi e a trovare le parole giuste, incomprensione del significato dei termini, conversazioni scarse o fuori tema. Nelle fasi più avanzate, il linguaggio può disorganizzarsi severamente fino a perdersi.
Movimenti e coordinazione: marcato deterioramento con lentezza motoria, andatura incerta/strascicata e equilibrio ridotto. Aumentano le cadute e la dipendenza nelle attività motorie quotidiane.
Deglutizione e nutrizione: nelle fasi severe si manifestano difficoltà a deglutire (disfagia), perdita di peso involontaria, rischio di aspirazione e conseguente polmonite; serve un attento monitoraggio dei pasti e, talvolta, valutazioni etiche sull’alimentazione assistita.
Diagnosi, biomarcatori e strumenti
La diagnosi è clinica e si basa su storia, osservazione del medico e racconto dei familiari/caregiver (spesso più affidabile dei resoconti del paziente, che può non riconoscere i propri deficit). Test neuropsicologici come il Mini-Mental State Examination sono ampiamente usati, insieme a batterie più complesse nelle fasi iniziali.
Neuroimmagini (TAC, RM) possono evidenziare segni di demenza ma non specificano sempre il tipo. Tecniche più avanzate includono PET dell’amiloide (ad es. PiB PET) e PET della tau, che visualizzano direttamente i depositi proteici; EEG e segnali elettrofisiologici sono allo studio per criteri aggiuntivi.
Liquor e sangue: l’analisi del liquido cefalorachidiano con Aβ e tau supporta il sospetto diagnostico; biomarcatori ematici emergenti possono indicare rischio o sostegno alla diagnosi, specialmente dopo l’esordio clinico. Test genetici non sono per tutti: si riservano a casi selezionati (esordio precoce o familiari).
Criteri NINCDS-ADRDA e linee guida aiutano a classificare il quadro come “possibile” o “probabile”; la certezza assoluta resta istologica (post mortem). La diagnosi differenziale richiede escludere cause reversibili e valutare depressione, comorbilità e altre demenze come la demenza di Parkinson. La diagnosi differenziale richiede escludere cause reversibili e valutare depressione e comorbilità.
Cause, ipotesi biologiche e fattori di rischio
La causa precisa non è definita. Tra le ipotesi: deficit colinergico, cascata amiloide (accumulo Aβ e tossicità), tauopatia con propagazione “transneurale” e perdita del trasporto assonale, disfunzione mitocondriale e neuroinfiammazione. È stata proposta un’impostazione “tipo diabete 3” per resistenza/deficit insulinico cerebrale.
Ipotesi infettive e ambientali: sono state segnalate correlazioni con infezioni fungine sistemiche, batteri come Porphyromonas gingivalis e presenza di citomegalovirus nel cervello; allo stato attuale, non vi è conferma conclusiva. Il ruolo dell’alluminio è controverso: molta evidenza lo smentisce, anche se alcuni lavori discutono associazioni fisiopatologiche.
Rischi vascolari e stili di vita: ipertensione, ipercolesterolemia, diabete, sedentarietà e fumo sono associati a maggior rischio e a decorso più rapido. La dieta mediterranea mostra potenziali proprietà protettive (anti-infiammatorie/antiossidanti); il ruolo di integratori come ginkgo biloba resta non supportato da evidenza solida.
Genetica: esordio precoce e tardivo
Le forme ad esordio precoce (rari casi sotto i 60 anni) sono spesso dovute a mutazioni autosomiche dominanti in APP, PSEN1 o PSEN2, con sovrapproduzione di Aβ; nella sindrome di Down (trisomia 21, sede del gene APP), i segni compaiono quasi universalmente intorno ai 40 anni.
Nell’esordio tardivo (la grande maggioranza dei casi), il fattore genetico di rischio più rilevante è APOE ε4, che favorisce la deposizione amiloide; tuttavia, non è deterministico. Ampi studi GWAS hanno identificato nuovi loci (per esempio HLA-DRA/DRB1, PTK2B, CLU, MS4A3, SCIMP, RABEP1, TNIP1, HAVCR2, TMEM106B, GRN, LILRA5, AGRN, NTN5), aprendo a nuove possibili terapie mirate.
Epigenetica e sesso: variazioni di metilazione (ad es. nel promotore FKBP5) si correlano con patologia tau; alcuni polimorfismi (come in RELN) potrebbero aumentare il rischio in donne. La genetica suggerisce un mosaico di fattori di rischio e protettivi che modulano età d’esordio e progressione.
Epidemiologia e impatto
Milioni di persone vivono con l’Alzheimer nel mondo (oltre 6 milioni negli USA, per lo più oltre i 65 anni). La prevalenza raddoppia circa ogni cinque anni dopo i 65 e cresce nettamente dopo gli 80; il rischio è più alto nelle donne, in particolare oltre gli 85 anni.
Incidenza e prevalenza aumentano con l’età: fasce 65–69 anni mostrano pochi nuovi casi per mille persone, che crescono progressivamente fino a valori molto elevati oltre i 90. Le stime globali indicano una crescente burden sanitario e sociale nelle prossime decadi.
Esordio precoce: meno del 5% di tutti i casi. Esordio tardivo: oltre il 95% dei casi, tipicamente dopo i 65 anni; l’età è il fattore di rischio maggiore, ma non l’unico determinante.
Trattamenti disponibili e linee di ricerca
Ad oggi non esiste una cura risolutiva. La terapia si basa su interventi non farmacologici e farmacologici. Gli inibitori dell’acetilcolinesterasi (donepezil, rivastigmina, galantamina) hanno effetti sintomatici moderati su apatia, iniziativa, allucinazioni e qualità di vita nelle fasi lievi-moderate; donepezil è usato anche in fase avanzata.
Memantina, antagonista dei recettori NMDA, è indicata nelle fasi moderate-avanzate, con benefici moderati e profilo di sicurezza accettabile; la combinazione con donepezil è statisticamente significativa ma clinicamente marginale. Effetti avversi comuni dei colinesterasici: nausea, vomito, crampi, bradicardia, perdita di appetito/peso; memantina può dare confusione, vertigini, cefalea.
Gestione dei disturbi comportamentali: gli antipsicotici si usano con grande cautela per aggressività/psicosi refrattarie, dato il rischio di eventi cerebrovascolari, effetti extrapiramidali e peggioramento cognitivo. L’obiettivo resta la riduzione del rischio e il comfort del paziente.
Ricerca: vaccini anti-amiloide hanno mostrato promesse precliniche ma anche eventi avversi in trial; studi su ultrasuoni focalizzati per aprire temporaneamente la barriera emato-encefalica e attivare la microglia sono incoraggianti nei modelli animali (miglioramento della memoria); citalopram ha ridotto la produzione di Aβ in piccoli studi; insulina intranasale e stimolazione cerebrale profonda sono linee esplorative ancora nelle prime fasi di evidenza clinica.
Interventi non farmacologici, ambiente e ruolo del caregiver
Gli interventi psicosociali includono approcci comportamentali, emozionali (validazione, reminiscenza, sostegno), cognitivi (orientamento alla realtà, training) e di stimolazione (arte, musica, attività assistite da animali, esercizio). Le evidenze sono eterogenee: alcuni benefici su umore, comportamento e funzioni quotidiane, ma spesso transitori e non sempre specifici per Alzheimer.
Ambiente e routine contano: stress familiare e cambi bruschi possono accelerare il declino; un contesto prevedibile e sereno, con esercizio e socialità, aiuta a rallentare la perdita funzionale. Misure pratiche: braccialetti con contatti, dispositivi GPS, etichette sugli oggetti, utensili adattati; il tutto per sicurezza e autonomia residua.
ADL e sicurezza: nelle fasi intermedie la persona può usare il bagno in autonomia ma necessita aiuto per compiti complessi (pagamenti, banca). Più avanti serve supporto per vestirsi, igiene, alimentazione, con attenzione a rischi di aspirazione. Le restrizioni fisiche sono raramente indicate e vanno limitate a situazioni di rischio immediato.
Complicanze mediche (piaghe da decubito, infezioni orali/urinarie/respiratorie, malnutrizione) richiedono prevenzione e supervisione medica. Nelle fasi terminali l’accento si sposta sulla qualità di vita, sul controllo dei sintomi e sul supporto al caregiver.
Prevenzione e stile di vita
Non esistono misure di prevenzione definitive, ma ridurre i fattori di rischio cardiovascolare (pressione, glicemia, colesterolo), mantenere attività fisica regolare e una dieta mediterranea possono associarsi a un minor rischio. L’uso a lungo termine di FANS è stato collegato in alcuni individui a minor probabilità, mentre statine e terapia ormonale sostitutiva non hanno mostrato efficacia preventiva.
Allenamento cognitivo e socialità (lettura, scacchi, cruciverba, interazioni frequenti), così come il bilinguismo, correlano con esordio più tardivo o minore gravità. Evidenze su campi magnetici, metalli (in particolare alluminio) e solventi sono contraddittorie e spesso di qualità metodologica limitata.
Riconoscere i segni tardivi e intervenire per tempo permette di ridurre rischi e complicanze, mantenendo dignità e comfort; una presa in carico integrata, attenta ai biomarcatori, alle comorbilità e al sostegno dei caregiver, consente di affrontare al meglio le sfide di una malattia complessa e ancora oggi incurabile.
