A Síndrome de Williams é uma condição genética rara que afeta cerca de 1 em cada 10.000 pessoas. Ela é causada pela deleção de um pequeno pedaço do cromossomo 7 e resulta em uma série de sintomas característicos, como atraso no desenvolvimento cognitivo, problemas cardíacos, características faciais distintas, hipercalcemia e dificuldades de aprendizagem.
O tratamento da Síndrome de Williams geralmente envolve uma abordagem multidisciplinar, com acompanhamento médico regular para monitorar problemas de saúde, terapia ocupacional e fisioterapia para ajudar no desenvolvimento físico e apoio educacional para lidar com as dificuldades de aprendizagem. Embora não haja cura para a síndrome, o tratamento adequado pode ajudar a melhorar a qualidade de vida e o bem-estar dos indivíduos afetados. É importante que as famílias e os cuidadores recebam apoio e orientação para lidar com os desafios únicos associados a essa condição genética.
Quais são as causas da síndrome de Williams?
A síndrome de Williams é uma condição genética rara que afeta aproximadamente 1 em cada 10.000 pessoas em todo o mundo. As causas dessa síndrome estão relacionadas a uma deleção (perda) de um pequeno pedaço do cromossomo 7. Essa deleção afeta diversos genes, incluindo o gene ELN, que é responsável pela produção da proteína elastina.
Essa perda do gene ELN resulta em problemas de desenvolvimento cardiovascular, como estenose da aorta e estenose pulmonar. Além disso, a deleção de outros genes afetados pela síndrome de Williams pode causar características faciais distintas, atraso no desenvolvimento cognitivo e problemas de saúde, como hipercalcemia e sensibilidade ao som.
Embora a síndrome de Williams seja causada por uma alteração genética, ela não é herdada dos pais. A deleção do cromossomo 7 geralmente ocorre de forma aleatória durante a formação dos espermatozoides ou óvulos. Portanto, é considerada uma condição genética esporádica.
O diagnóstico da síndrome de Williams é feito por meio de testes genéticos que identificam a deleção no cromossomo 7. Uma vez diagnosticada, o tratamento da síndrome de Williams é multidisciplinar, envolvendo acompanhamento médico, terapias ocupacionais e fonoaudiologia para ajudar a lidar com os sintomas e melhorar a qualidade de vida do paciente.
Característica principal de crianças com Síndrome de William: alegria e sociabilidade excepcionais.
A Síndrome de Williams, também conhecida como Síndrome de Williams-Beuren, é uma condição genética rara que afeta cerca de 1 em cada 10.000 pessoas em todo o mundo. Ela é causada por uma deleção de genes no cromossomo 7, resultando em uma série de sintomas característicos.
Uma das características mais marcantes da Síndrome de Williams é a alegria e sociabilidade excepcionais das crianças afetadas. Elas tendem a ser extremamente amigáveis, extrovertidas e carinhosas, buscando constantemente interação social e contato físico com os outros.
Além da sociabilidade, as crianças com Síndrome de Williams também podem apresentar outros sintomas, como deficiência intelectual leve a moderada, dificuldades de aprendizagem, problemas cardíacos congênitos, hiperacusia (sensibilidade auditiva aumentada) e rostos característicos com olhos grandes e lábios cheios.
O tratamento para a Síndrome de Williams envolve uma abordagem multidisciplinar, com acompanhamento médico regular, terapia ocupacional, terapia da fala e educação especializada. O objetivo é ajudar as crianças a desenvolver suas habilidades e alcançar seu pleno potencial, garantindo uma melhor qualidade de vida.
Apesar dos desafios que a Síndrome de Williams pode trazer, as crianças afetadas continuam a surpreender com sua alegria contagiante e capacidade de criar laços afetivos profundos com aqueles ao seu redor. Com o apoio adequado, elas podem levar uma vida feliz e realizada, contribuindo de maneira única para o mundo ao seu redor.
Identificando os sinais da síndrome de Williams em crianças: guia prático e informativo.
A Síndrome de Williams é uma condição genética rara que afeta cerca de 1 em cada 10.000 pessoas em todo o mundo. É causada por uma deleção no cromossomo 7, resultando em diversos sintomas que podem ser identificados em crianças desde cedo.
Alguns dos sinais mais comuns da Síndrome de Williams incluem facial características distintas, como um nariz pequeno e achatado, lábios cheios, olhos brilhantes e uma testa larga. Além disso, as crianças com a síndrome tendem a ter um baixo peso ao nascer e dificuldades no ganho de peso, além de apresentarem um atraso no desenvolvimento motor e cognitivo.
Outros sintomas que podem ser observados incluem problemas cardíacos, como estenose aórtica e supravalvular pulmonar, dificuldades de alimentação devido a problemas de sucção e deglutição, além de um padrão de comportamento social extrovertido e amigável.
O diagnóstico da Síndrome de Williams é feito por meio de testes genéticos, que identificam a deleção no cromossomo 7. Uma vez diagnosticada, o tratamento da síndrome envolve uma abordagem multidisciplinar, com acompanhamento médico, terapia ocupacional, terapia da fala e terapia comportamental.
Com o devido suporte e cuidado, as crianças com essa condição podem levar uma vida plena e feliz.
Estratégias para lidar com crianças com síndrome de Williams: dicas úteis e eficazes.
A Síndrome de Williams é uma condição genética rara que afeta o desenvolvimento físico e cognitivo das crianças. Caracteriza-se por sintomas como rosto em forma de coração, atrasos no desenvolvimento, dificuldades de aprendizagem e habilidades sociais incomuns. Apesar dos desafios que essa síndrome pode trazer, existem estratégias eficazes para lidar com crianças com Williams.
Uma dica importante é estabelecer rotinas claras e previsíveis para a criança. Isso ajuda a diminuir a ansiedade e melhora a sua capacidade de se adaptar a diferentes situações. Além disso, incentivar a comunicação é essencial. Use linguagem simples e direta, e dê tempo para que a criança processe as informações e responda.
Estimular atividades sensoriais também pode ser benéfico. Brinquedos texturizados, música e atividades que envolvam os sentidos podem ajudar a criança a se sentir mais confortável e engajada. Além disso, é importante promover a interação social. Incentive a criança a participar de atividades em grupo e a desenvolver habilidades sociais.
No entanto, é fundamental lembrar que cada criança é única e pode responder de maneira diferente a essas estratégias. Por isso, é importante observar e adaptar as abordagens de acordo com as necessidades específicas de cada criança com Síndrome de Williams.
Síndrome de Williams: sintomas, causas, tratamento
A Síndrome de Williams é uma desordem de desenvolvimento de origem genética que está associado com um perfil característico de afectação física e cognitiva.Especificamente no nível clínico, é caracterizado por 4 pontos cardeais: 1) características e características faciais atípicas, 2) atraso generalizado do desenvolvimento psicomotor e perfil neurocognitivo específico, 3) alterações cardiovasculares et) possibilidade de desenvolvimento de hipercalcemia infantil.
Embora a síndrome de Williams seja considerada uma doença rara, existem milhares de pessoas afetadas em todo o mundo.Quanto ao diagnóstico, o exame clínico geralmente fornece os achados necessários para o seu estabelecimento; no entanto, para descartar outras patologias e falsos positivos, um estudo genético geralmente é iniciado por meio de várias técnicas.
Por outro lado, não existe uma cura para a síndrome de Williams nem um protocolo de tratamento padrão; muitas das intervenções terapêuticas tentarão regular as complicações médicas.Além disso, será essencial incluir programas de atendimento precoce, educação especial individualizada e estímulo neuropsicológico nas intervenções.
Características da síndrome de Williams
A síndrome de Williams é um distúrbio do desenvolvimento que pode afetar significativamente diferentes áreas.
Geralmente, essa patologia é caracterizada pela presença de características faciais atípicas ou distúrbios cardiovasculares, incapacidade intelectual moderada, problemas de aprendizado e traços distintos de personalidade .
Assim, o primeiro paciente com síndrome de Williams foi descrito pelo Dr. Guido Fanconi, em um relatório clínico de 1952. No entanto, foi o cardiologista Joseph Williams que, em 1961, identificou com precisão essa patologia, enquanto foi descrito pelo Beuren alemão
Por esse motivo, a síndrome de Williams recebe o nome de ambos os autores (síndrome de Williams-Beuren) ou simplesmente do primeiro.
Embora, até alguns anos atrás, a patologia tenha sido identificada com base em características fenotípicas, em 1993, Edward et al. Encontraram uma anomalia genética no cromossomo 7q 11.23 como causa etiológica.
Embora a síndrome de Williams sofra da presença de uma ampla variedade de complicações médicas secundárias, ela não apresenta alta taxa de mortalidade. Em muitos casos, os indivíduos afetados são capazes de atingir um nível funcional independente.
Estatisticas
A síndrome de Williams é considerada um distúrbio genético raro ou raro.
A Associação da Síndrome de Williams, entre outras instituições, estimou que a síndrome de Williams tem uma prevalência de aproximadamente 1 caso por 10.000 pessoas em todo o mundo.Especificamente, foi identificado que pode haver cerca de 20.000 ou 30.000 afetados nos Estados Unidos.
Em relação à distribuição da patologia por sexo, não existem dados recentes que indiquem uma prevalência mais alta em nenhuma delas; além disso, não foram identificadas diferenças entre regiões geográficas ou grupos étnicos.
Por outro lado, também sabemos que a síndrome de Williams é uma condição médica esporádica, embora alguns casos de transmissão familiar tenham sido descritos .
Signos e sintomas
A síndrome de Williams, como outras patologias de origem genética, apresenta um curso clínico caracterizado por um envolvimento multissistêmico.
Muitos autores, como González Fernández e Uyaguari Quezada, descrevem o espectro clínico da síndrome de Williams categorizado em várias áreas: características biomédicas, características psicomotoras e cognitivas, características psicológicas e comportamentais, entre outras.
Características biomédicas
O envolvimento físico presente na síndrome de Wiliams é diverso, dentre os achados clínicos mais frequentes que podemos observar :
Atraso generalizado de crescimento
Já durante a gravidez, um desenvolvimento retardado ou retardado pode ser detectado. As crianças afetadas pela síndrome de Williams geralmente nascem com peso e altura reduzidos. Além disso, uma vez atingida a fase adulta, a altura total é geralmente menor do que a da população em geral, aproximadamente 10 a 15 cm.
Características faciais atípicas
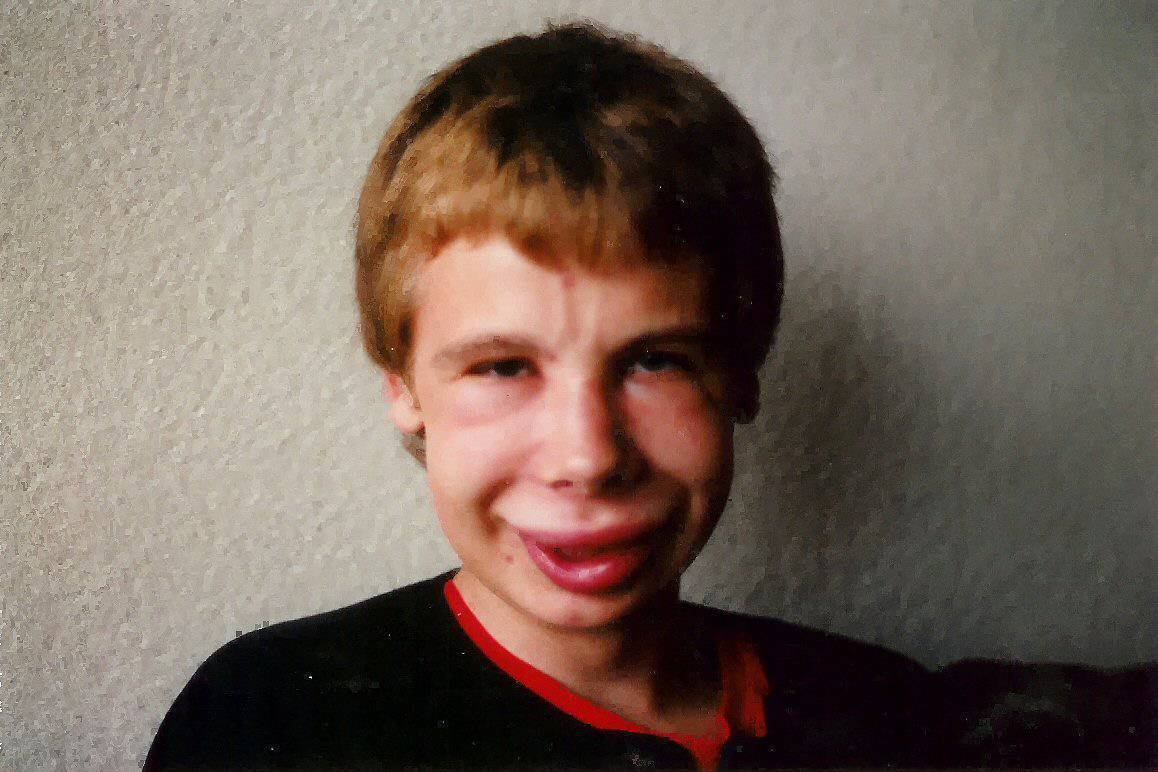
As alterações faciais são um dos achados clínicos mais característicos dessa síndrome. Nos indivíduos afetados, podemos observar uma testa significativamente estreita, dobras cutâneas acentuadas na fenda palpebral, estrabismo, íris estrelada, nariz curto e achatado, maçãs do rosto proeminentes e queixo menor que o normal.
Distúrbios músculo-esqueléticos
No caso de alterações relacionadas ao desenvolvimento de músculos e ossos, é possível observar presença de tônus e força muscular reduzidos, flacidez articular, escoliose, contraturas, entre outros. No nível visual, pode-se observar uma postura caracterizada por ombros caídos e membros inferiores semi-flexionados.
Aparelhos auditivos prejudicados
Embora em geral não haja anormalidades ou malformações significativas no pavilhão auditivo, em todos os casos ocorre um aumento na sensibilidade auditiva. Os indivíduos afetados tendem a perceber ou experimentar certos sons como irritantes ou dolorosos.
Distúrbios da pele
A pele geralmente tem pouca elasticidade, portanto é possível observar sinais precoces de envelhecimento.Além disso, é possível o desenvolvimento de hérnias, especialmente na região inguinal e umbilical.
Distúrbios cardiovasculares
As diferentes anormalidades no coração e nos vasos sanguíneos constituem a complicação médica mais significativa, pois podem comprometer a sobrevivência da pessoa afetada.
Entre as anomalias cardiovasculares, algumas das mais comuns são estenose aórtica supravalvar, estenose de ramo pulmonar, estenose da válvula aórtica. Todas essas alterações, no nível clínico, podem afetar outros territórios vasculares e até o cérebro, devido ao desenvolvimento da hipertensão arterial.
Alterações do aparelho geniturinário
Anormalidades relacionadas à função renal e bexiga são muito comuns. Além disso, também pode ser detectado acúmulo de cálcio (nefrocalcinose), urgência urinária ou enurese noturna.
Características psicomotoras e cognitivas
No nível cognitivo, as características mais significativas são constituídas por um atraso geral na aquisição de habilidades motoras, retardo intelectual moderado e várias alterações relacionadas à percepção visual.
Alterações psicomotoras
São descritas várias alterações relacionadas ao equilíbrio, problemas de coordenação, principalmente devido à presença de anomalias osteomusculares e que causarão, entre outras coisas, atraso na aquisição da marcha, habilidades motoras finais, etc.
Características cognitivas
É possível encontrar um retardo mental moderado, o QI típico das pessoas afetadas geralmente varia entre 60 e 70. Quanto às áreas específicas afetadas, há uma clara assimetria: além da coordenação psicomotora, percepção visual e integração, geralmente ser claramente afetado, enquanto áreas como a linguagem tendem a ser mais desenvolvidas.
Características linguísticas
Nos estágios mais iniciais , geralmente ocorre um atraso na aquisição de habilidades linguísticas, no entanto, geralmente se recupera em torno de 3-4 anos. As crianças com síndrome de Williams, geralmente têm boa comunicação expressiva, são capazes de usar vocabulário contextualizado, gramática correta, contato visual, expressões faciais etc.
Características psicológicas e comportamentais
Um dos achados mais significativos da síndrome de Williams é o excepcional comportamento social das pessoas afetadas. Embora em alguns casos possa haver crises ansiosas ou preocupações excessivas, elas são muito empáticas e sensíveis.
Causas
Pesquisas mais recentes indicaram que a causa da síndrome de Williams é encontrada em várias alterações genéticas no cromossomo 7. Os cromossomos carregam as informações genéticas de cada pessoa e estão localizados no núcleo das células do corpo.
Nos seres humanos, podemos encontrar 46 cromossomos distribuídos em pares. Eles são numerados de 1 a 23, exceto o último casal constituído pelos cromossomos sexuais, chamado XX no caso de mulheres XY no caso de homens. Assim, dentro de cada cromossomo pode haver inúmeros genes.
Especificamente, o processo anômalo identificado na síndrome de Williams é uma microceleção ou ruptura de uma molécula de DNA que confirma esse cromossomo. Normalmente, esse tipo de erro ocorre na fase de desenvolvimento dos gametas masculinos ou femininos.
As anomalias genéticas estão na área 7q11.23, na qual foram identificados mais de 25 genes diferentes relacionados ao padrão clínico característico dessa patologia.
Alguns dos genes, como Clip2, ELN, GTF21, GTF2IRD1 ou LIMK1, estão ausentes nos afetados. A perda de ELN está relacionada ao tecido conjuntivo, pele e anormalidades cardiovasculares.
Por outro lado, algumas pesquisas indicam que a perda dos genes Clip2, GTF2I, GTF2IRD1 e LIMK1 pode explicar as alterações nos processos visuoperceptivos, o fenótipo comportamental ou os déficits cognitivos.
Além disso, especificamente, o gene GTF2IRD1 parece ter um papel proeminente no desenvolvimento de características faciais atípicas. Por seu lado, o gene NCF1 parece estar relacionado a um alto risco de desenvolver hipertensão.
Diagnóstico
Até os últimos anos, o diagnóstico da síndrome de Williams era realizado exclusivamente com base na observação das características fenotípicas (alterações faciais, incapacidade intelectual, déficits cognitivos específicos, entre outras).
No entanto, atualmente, o diagnóstico da síndrome de Williams é geralmente realizado em dois momentos: análise de achados clínicos e estudos de confirmação genética .Assim, o diagnóstico clínico geralmente inclui:
– Exploração e avaliação física e neurológica.
– Análise de parâmetros de crescimento.
– Exploração do sistema cardiorrespiratório.
– Exploração nefrourológica.
– Análise dos níveis de cálcio na urina e no sangue.
– análise oftalmológica.
Por outro lado, a análise genética é utilizada para confirmar a presença de alterações genéticas compatíveis com a síndrome de Williams, dentre os testes mais comuns está a técnica de hibridização fluorescente in situ (FIHS).
Após a extração de uma amostra de sangue, a técnica de hibridação in situ é realizada pela marcação de sondas AND que são detectadas sob uma luz fluorescente.
Tratamento
Não existe tratamento específico para a síndrome de Williams; no entanto, essa patologia está associada a múltiplas complicações em diferentes órgãos, portanto as intervenções médicas serão direcionadas ao tratamento delas.
Os autores González Fernández e Uyaguari Quezada enfatizam que todas as intervenções devem ter um caráter multidisciplinar marcado, o que permite o tratamento da variedade sintomática característica dessa síndrome.Além disso, eles também indicam várias medidas terapêuticas, dependendo da área afetada:
Área médica
Nesse caso, complicações médicas, como anormalidades cardíacas ou malformações músculo-esqueléticas, geralmente requerem tratamento baseado principalmente na administração de medicamentos e procedimentos cirúrgicos. Profissionais médicos de diferentes áreas (pediatras, cardiologistas, oftalmologistas etc.) geralmente participam do tratamento de sintomas físicos.
Área neuropsicológica
Déficits cognitivos, como comprometimento visuoperceptivo ou atraso lingüístico, devem ser abordados precocemente. A estimulação cognitiva e a reabilitação serão um fator determinante para alcançar uma vida autônoma durante a vida adulta.
Área psicológica
Embora os afetados pela síndrome de Williams geralmente tenham um bom funcionamento social, às vezes eles precisam mostrar comportamentos excessivamente ansiosos e desenvolver comportamentos perseverantes ou fobias.
Portanto, nesses casos, será essencial iniciar uma abordagem psicológica, por meio de várias estratégias eficazes para minimizar esses problemas ou dificuldades.
Referências
- Antonell, A., del Campo, M., Flores, R., Campuzano, V. e Pérez-Jurado, L. (2006). Síndrome de Willims: aspectos clínicos e bases moleculares. Rev Neurol, 69-75.
- Cleveland Clinic (2013). Síndrome de Williams Obtido na Cleveland Clinic.
- del Campo Castenelles, M., & Pérez Jurado, L. (2010). Protocolo de acompanhamento na síndrome de Williams. Associação Espanhola de Pediatria, 116-124.
- Galaburda, A., Holinger, D., Mills, D., Reiss, A., Korenberg, J. e Bellugui, U. (2003). Síndrome de Williams. Um resumo dos achados cognitivos, eletrofisiológicos, anatomofuncionais, microanatóticos e genéticos. Rev Neurol, 132-137.
- García-Nonell, C., Rigau-Ratera, E., Artigas-Pallarés, J., García Sánchez, C., e Estévez-González, A. (2003). Síndrome de Williams: memória, funções visuoespaciais e funções visuoconstrutivas. Rev Neurol, 826-830.
- Orphanet (2006). Síndrome de Williams Obtido da Orphanet.
- Associação da Síndrome de Williams. (2016). O QUE É SÍNDROME DE WILLIAMS? Obtido na Williams Syndrome Association.