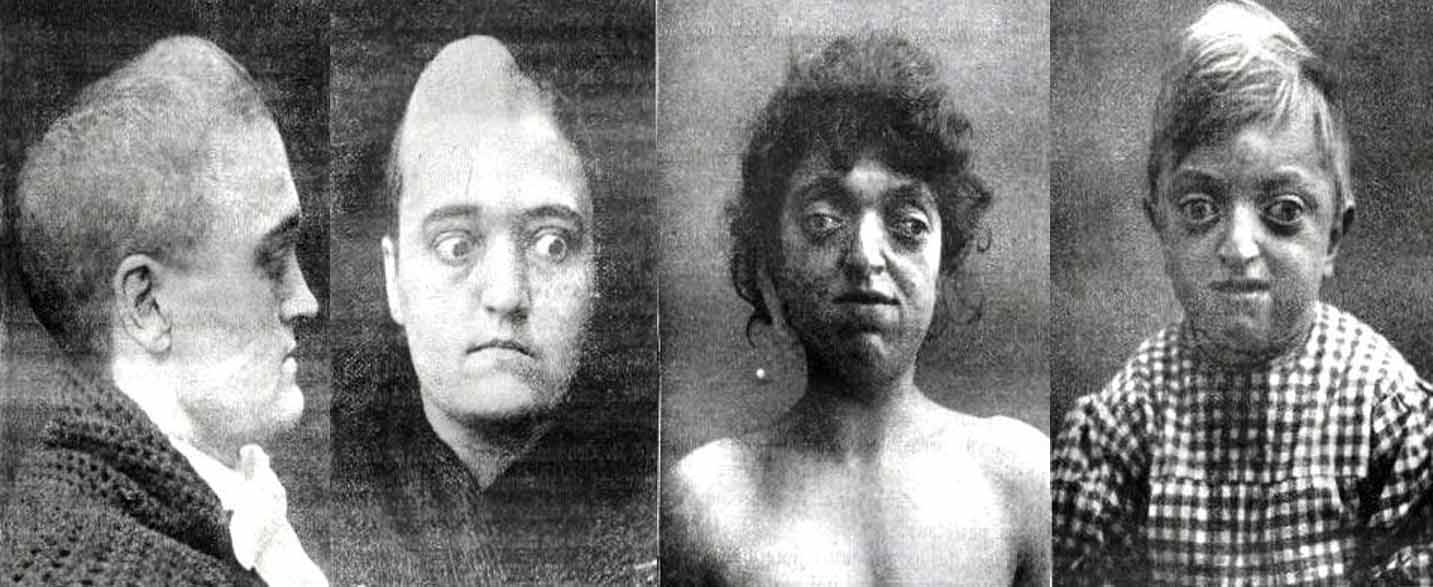
A Síndrome de Crouzon é uma condição genética rara que afeta o desenvolvimento do crânio e do rosto. Os principais sintomas incluem uma aparência facial distinta, com olhos protuberantes, nariz largo e boca pequena, além de problemas dentários e auditivos. As causas da síndrome estão relacionadas a mutações genéticas que afetam o desenvolvimento dos ossos do crânio. O tratamento geralmente envolve cirurgias corretivas para melhorar a função e a estética do rosto e do crânio, além de acompanhamento médico especializado ao longo da vida. É importante um diagnóstico precoce e um plano de tratamento individualizado para melhorar a qualidade de vida dos pacientes com Síndrome de Crouzon.
Quais são as causas da síndrome de Crouzon?
A síndrome de Crouzon é uma condição genética rara que afeta o desenvolvimento dos ossos do crânio e do rosto. As causas desta síndrome estão relacionadas a mutações genéticas no gene FGFR2, que é responsável por controlar o crescimento e desenvolvimento das células ósseas. Estas mutações causam uma superprodução de proteínas que interferem na formação adequada dos ossos do crânio e do rosto, resultando em características faciais distintas.
As mutações genéticas que causam a síndrome de Crouzon podem ser herdadas dos pais ou ocorrerem de forma espontânea. Estas mutações afetam o modo como as células ósseas se desenvolvem durante a gestação, resultando em uma fusão prematura das suturas cranianas e um crescimento anormal dos ossos da face. Isso leva a uma aparência facial característica, com olhos proeminentes, testa estreita e nariz pequeno.
É importante ressaltar que a síndrome de Crouzon não é causada por fatores ambientais ou pela forma como a gestação é conduzida. Trata-se de uma condição genética que pode ser diagnosticada por meio de exames genéticos e que requer acompanhamento médico especializado ao longo da vida. O tratamento para a síndrome de Crouzon envolve intervenções cirúrgicas para corrigir as deformidades faciais e garantir um desenvolvimento adequado do crânio e do rosto.
Significado da disostose craniofacial: uma condição genética que afeta o desenvolvimento dos ossos do crânio e face.
Significado da disostose craniofacial: uma condição genética que afeta o desenvolvimento dos ossos do crânio e face. Esta condição resulta em uma formação anormal dos ossos da cabeça, afetando a forma e o tamanho do crânio e da face. A disostose craniofacial pode causar diferentes problemas de saúde e estética, dependendo da gravidade do quadro.
Síndrome de Crouzon: sintomas, causas, tratamento
A Síndrome de Crouzon é uma forma específica de disostose craniofacial, caracterizada por sintomas como órbitas oculares proeminentes, maxilar superior subdesenvolvido e alterações na forma do crânio. Esta síndrome é causada por uma mutação genética e pode variar em gravidade de um paciente para outro.
Os sintomas da Síndrome de Crouzon podem incluir problemas de visão, dificuldade para respirar, apneia do sono, dores de cabeça frequentes, entre outros. O diagnóstico é feito com base em exames clínicos e de imagem, e o tratamento geralmente envolve cirurgias para corrigir as deformidades craniofaciais e melhorar a qualidade de vida do paciente.
É importante ressaltar que o tratamento da Síndrome de Crouzon deve ser individualizado, levando em consideração as necessidades específicas de cada paciente. Além das cirurgias corretivas, podem ser necessárias terapias complementares e acompanhamento médico regular para garantir o bem-estar do paciente a longo prazo.
Origem e fatores desencadeantes da síndrome de Apert: o que sabemos até o momento.
A Síndrome de Apert é uma condição genética rara que afeta o desenvolvimento craniofacial e esquelético. Ela é causada por mutações no gene FGFR2, que é responsável pela regulação do crescimento celular e da formação de ossos. Essas mutações ocorrem de forma espontânea, sem uma causa específica conhecida.
Os principais fatores desencadeantes da Síndrome de Apert incluem a idade avançada dos pais no momento da concepção, o que aumenta o risco de mutações genéticas, e a exposição a substâncias tóxicas durante a gravidez. Além disso, existem evidências de que a síndrome possa ter um componente hereditário em alguns casos.
Síndrome de Crouzon: sintomas, causas, tratamento
A Síndrome de Crouzon é uma condição genética que também afeta o desenvolvimento craniofacial, mas de forma diferente da Síndrome de Apert. Os principais sintomas incluem craniossinostose, que é o fechamento prematuro das suturas cranianas, exoftalmia, que é a protrusão dos olhos, e hipoplasia maxilar, que é o subdesenvolvimento da mandíbula.
As causas da Síndrome de Crouzon são semelhantes às da Síndrome de Apert, com mutações genéticas no gene FGFR2 sendo as principais responsáveis pela condição. O tratamento da Síndrome de Crouzon envolve cirurgias corretivas para corrigir as deformidades craniofaciais e melhorar a função respiratória e visual dos pacientes.
Entenda o que é a síndrome de Hurler e seus sintomas característicos.
A Síndrome de Hurler, também conhecida como mucopolissacaridose tipo I, é uma doença genética rara que afeta o metabolismo dos mucopolissacarídeos. Esta condição resulta na acumulação desses compostos nas células do corpo, levando a diversos problemas de saúde.
Os sintomas característicos da Síndrome de Hurler incluem deformidades ósseas, como o crânio alongado e achatado, além de problemas de visão e audição. Outros sinais incluem baixa estatura, língua grande, hérnia umbilical, problemas cardíacos e respiratórios, além de atrasos no desenvolvimento.
O diagnóstico da Síndrome de Hurler é feito através de exames clínicos, testes genéticos e análise dos níveis de enzimas. Infelizmente, não há cura para esta condição, mas o tratamento visa aliviar os sintomas e melhorar a qualidade de vida do paciente. Isso pode incluir terapias de reposição enzimática, cirurgias corretivas, fisioterapia e acompanhamento médico regular.
Síndrome de Crouzon: sintomas, causas, tratamento
A síndrome de Crouzon é um produto craniofacial malformação de um fecho ou desenvolvimento anormal de suturas cranianas e, portanto, produz várias anomalias em face e do crânio.É uma patologia de origem congênita ligada à presença de uma mutação parcial ou completa do gene FGFR2, relacionada ao fator de crescimento de fibroblastos (FGFR).
No nível clínico, a síndrome de Crouzon é caracterizada pela presença de abaulamento ou abaulamento da parte frontal do crânio, encurtamento do volume total da cabeça, hipoplasia maxilar ou desenvolvimento normal das órbitas oculares, entre outros aspectos.
Quanto ao diagnóstico, os sinais clínicos geralmente não são claramente visíveis ao nascimento. Geralmente, as características físicas tendem a se manifestar aproximadamente aos dois anos de idade. Assim, o diagnóstico é confirmado com base em um exame físico detalhado e em um estudo genético.
Embora não exista cura para a síndrome de Crouzon, existe uma grande variedade de abordagens terapêuticas que podem melhorar significativamente as complicações médicas decorrentes dessa patologia.
Em todos os casos, o tratamento de escolha é baseado no trabalho de uma equipe multidisciplinar: odontologia, neurocirurgia, oftalmologia, traumatologia, fisioterapia, fonoaudiologia, neuropsicologia, etc.
Características da síndrome de Crouzon

Especificamente, essa patologia foi descrita inicialmente em 1912 pelo cirurgião de origem francesa Octavie Crouzon.Já nos primeiros casos clínicos descritos na literatura médica e experimental, foi possível encontrar uma associação explícita dos sinais craniofaciais a uma formação anormal das suturas cranianas (Beltrán, Rosas e Jorges, X).
As declarações mais atuais dessa patologia o definem como um produto de distúrbio genético de uma craniossinose ou fechamento precoce dos ossos que compõem o crânio.
A configuração do crânio durante a fase infantil ou de desenvolvimento, possui uma estrutura oval, sendo mais ampla pela área posterior. Assim, as peças ósseas (occipital, temporal, parietal e frontal) são geralmente formadas no quinto mês de gravidez e são unidas por um tecido conjuntivo ou fibroso, as suturas cranianas.
As suturas cranianas, portanto, permitem o crescimento do volume da cabeça e do cérebro, graças à sua flexibilidade. Além disso, seu fechamento começa a se desenvolver progressivamente entre 9 e 24 meses.
Quando ocorre uma alteração desse processo, como a craniostenose, ocorre um fechamento precoce dessas estruturas fibrosas.
Dessa maneira, esse evento impede que a estrutura que compõe o crânio, a face e o cérebro se forme normalmente. Como conseqüência, a pessoa afetada desenvolverá várias malformações que afetam os olhos, a posição da mandíbula, a forma do nariz, os dentes ou a formação dos lábios e palato.
Embora a maioria dos indivíduos com síndrome de Crouzon tenha falta de moradia normal ou esperada para sua faixa etária, o desenvolvimento cerebral habitual pode ser mais lento. Como resultado, podem surgir várias dificuldades de aprendizado que, juntamente com anomalias dentárias e maxilares, retardarão significativamente a aquisição da linguagem.
Além do termo mais comumente usado, síndrome de Crouzon, essa patologia também pode ser referenciada com outro tipo de denominação: craniostenose de Crouzon, disostose craniofacial ou disostose craniofacial de Crouzon (National Organization for Rare Disorders, 2007).
Estatisticas

A frequência da síndrome de Crouzon foi estimada em aproximadamente 16 casos por milhão de recém-nascidos em todo o mundo.Mais especificamente, o Hospital Seattle Chindre (2016) aponta que a síndrome de Crouzon é uma patologia que pode ocorrer em 1,6% das pessoas em cada 100.000.
Além disso, é uma das patologias mais frequentes derivadas da craniossinose. Aproximadamente 4,5% das pessoas que tiveram craniossinose têm síndrome de Crouzon.
Por outro lado, em termos de prevalência por diferença de sexo, não foram encontrados dados estatísticos que indiquem um aumento significativo no número de casos em nenhum deles. Além disso, a ocorrência da síndrome de Crouzon não foi associada a regiões geográficas específicas ou a grupos étnicos específicos.
Signos e sintomas

As características clínicas e as complicações médicas típicas da síndrome de Crouzon podem variar significativamente entre os indivíduos afetados. No entanto, o achado cardinal em todos é a presença de craniossinostose.
Craniossinostose
Autores como Sanahuja et al., (2012) definem a craniossinostose como um evento patológico que resulta na fusão precoce de uma ou várias suturas cranianas.
Dessa forma, o desenvolvimento do crânio é deformado pelo crescimento em uma direção paralela às áreas afetadas, ou seja, o crescimento diminui nas suturas fundidas e continua progressivamente nas abertas.
Na síndrome de Crouzon, o fechamento das placas ósseas cranianas ocorre 2 ou 3 anos antes do nascimento, no entanto, em outros casos, pode ser evidente ao nascimento.
Além disso, o grau de envolvimento pode ser variável, dependendo das áreas ou suturas afetadas pela fusão.
Nos casos mais graves, é possível observar uma fusão das suturas das peças ósseas que formam a testa e os lados superiores do crânio, ou seja , as suturas coronal e sagital, por um lado, e as suturas parietais, por outro. Além disso, em outros casos, também é possível detectar uma sutura das estruturas ósseas mais posteriores.
Assim, a craniossinostose é o evento etiológico que dá origem ao restante dos sintomas e complicações médicas da síndrome de Crouzon.
Malformações cranianas
A fusão das suturas cranianas pode levar a um amplo padrão de anomalias e malformações cranianas, dentre as mais comuns:
– Braquicefalia: é possível observar uma alteração na estrutura da cabeça, mostrando um comprimento reduzido, aumentando a largura e o achatamento das áreas posterior e occipital.
– Escafocefalia: em outros casos, observaremos uma cabeça de forma alongada e estreita. As áreas mais frontais crescem para a frente e para cima, enquanto nas áreas occipitais é possível observar uma forma ondulada ou de pico.
– Trigonocefalia: neste caso, a cabeça mostra uma deformidade em forma de triângulo, com uma protuberância significativa da testa e uma posição próxima dos dois olhos.
– Craniossinotose do tipo crânio ou trevo ou Keeblattschadel: essa alteração constitui uma síndrome específica, na qual a cabeça adquire a forma de trevo. Especificamente, uma proeminência bilateral das áreas temporais e da parte superior da cabeça pode ser observada.
Distúrbios oculares
A área oftalmológica é uma das mais afetadas na síndrome de Crouzon, algumas das patologias mais comuns podem incluir:
– Proptose: a estrutura óssea das órbitas oculares desenvolve-se com profundidade rasa e, consequentemente, os globos oculares têm uma posição avançada, ou seja, parecem sobressair dessas cavidades.
– Ceratite de exposição: a posição anormal dos globos oculares resulta em maior exposição de suas estruturas; portanto, o desenvolvimento de uma inflamação significativa dessas estruturas oculares localizadas nas áreas mais avançadas.
– Conjuntivite: como no caso anterior, a exposição das estruturas oculares, pode causar o desenvolvimento de infecções, como a conjuntivite, que causa inflamação dos tecidos conjuntivos.
– Hipertelorismo ocular: em alguns indivíduos, é possível observar um aumento significativo na distância entre os dois olhos.
– Estrabismo divergente ou exotropia: neste caso, é possível observar uma ausência de simetria ou paralelismo entre os dois olhos, ou seja, quando um ou ambos os olhos se desviam para as áreas laterais.
– Atrofia óptica : também pode ocorrer o desenvolvimento de uma degeneração progressiva dos terminais nervosos responsáveis pela transmissão de informações visuais das áreas oculares para o cérebro.
– Nistagmo: alguns indivíduos apresentam persistentemente movimentos oculares involuntários, com apresentação arrítmica e rápida.
– Cataratas: neste caso, a lente do olho torna-se opaca e, portanto, dificulta a passagem da luz para a terina, para processamento. Os indivíduos afetados terão uma deterioração significativa em sua capacidade visual.
– Coloboma da íris: pode aparecer uma ausência parcial ou total da íris , ou seja, a área colorida do olho.
– Deficiência visual: boa parte das pessoas afetadas tem uma deterioração significativa da capacidade visual; em muitos casos, pode ocorrer na forma de cegueira com gravidade variável.
Malformações faciais
– Protuberância frontal: uma das características mais características da síndrome de Crouzon é a presença de uma testa protuberante ou proeminente. A estrutura óssea frontal tende a crescer anormalmente para a frente.
– Malformação nasal: em alguns casos, é possível observar um nariz em forma de “bico de papagaio”, ou seja, com a ponta nasal para baixo ou para baixo.
– Hipoplasia do terço facial médio: neste caso, há um desenvolvimento parcial ou mais lento das áreas centrais da face.
Malformações orais e maxilares
– Hipoplasia maxilar: em boa parte dos indivíduos, apresenta mandíbula superior pequena ou não desenvolvida.
– prognatismo mandibular: essa patologia é caracterizada por uma proeminência ou tendência a deixar a mandíbula inferior, ou seja, é posicionada em uma posição mais avançada que a superior.
– Fenda palatina: em alguns casos, é possível observar um fechamento incompleto do teto do palato, incluindo a estrutura labial.
– Má oclusão dentária: o desalinhamento das peças dentárias ou a alteração da posição da mordida é um dos achados maxilares e orais mais frequentes.
Distúrbios neurológicos e neuropsicológicos
As malformações cranianas podem impedir o crescimento normal e exponencial das estruturas cerebrais e, portanto, levar à presença variável de várias anormalidades, como:
– Dores de cabeça e dores de cabeça recorrentes.
– episódios de apreensão.
– Atraso mental.
– Hidrocefalia progressiva.
– Pressão intracraniana aumentada.
Causas
A origem genética da síndrome de Crouzon está associada a uma mutação específica do gene FGFR2.Especificamente, esse gene tem a função essencial de fornecer as instruções necessárias para o desenvolvimento da produção do fator de crescimento de fibroblastos.
Entre outras coisas, eles são responsáveis por sinalizar para células imaturas sua conversão ou diferenciação em células ósseas, durante o estágio de desenvolvimento embrionário.
No caso da síndrome de Crouzon, os especialistas propõem um aumento ou superestimação da sinalização pela proteína FGFR2 e , consequentemente, os ossos do crânio precisam se fundir prematuramente.
Embora a principal mutação tenha sido identificada no gene FGFR2 localizado no cromossomo 10, alguns relatos clínicos associaram o curso clínico dessa patologia a uma mutação do gene FGFR3 no cromossomo 4.
Diagnóstico
Como apontamos, a maioria das pessoas afetadas começa a desenvolver características físicas óbvias durante o estágio infantil, geralmente após os 2 anos de idade. Existem poucos casos em que os sinais e sintomas mais característicos são diretamente observáveis no nascimento.
Geralmente, o passo inicial da síndrome de Crouzons é baseado principalmente na identificação de características clínicas craniofaciais. Além disso, para confirmar certas características ou anormalidades ósseas, vários exames laboratoriais podem ser utilizados: radiografias tradicionais, tomografia axial computadorizada , biópsia de pele etc.
Além disso, estudos genéticos são essenciais para determinar a presença de mutações genéticas e identificar um possível padrão herdado.
Tratamento
Atualmente, estudos experimentais falharam em identificar qualquer tipo de terapia que diminua a fusão craniana. Portanto, as intervenções são orientadas principalmente para controle e gestão sintomática.
As equipes responsáveis pelo tratamento dessa patologia são geralmente formadas por especialistas de diversas áreas: cirurgia, pediatria, fisioterapia, fonoaudiologia, psicologia, neuropsicologia, etc.
Graças aos atuais avanços em procedimentos e ferramentas cirúrgicas, muitas das malformações craniofaciais podem ser corrigidas com uma alta taxa de sucesso.
Referências
- AAMADE (2012). Síndrome de Crouzon. Obtido da Associação de Anormalidades e Malformações Dentofaciais.
- Beltrán, R., Rosas, N., & Jorges, I. (2016). Síndrome de Crouzon. Revista Neurology.
- Hospital Infantil de Boston. (2016). Síndrome de Crouzon em crianças. Obtido no Hospital Infantil de Boston.
- Associação craniofacial infantil. (2016). Guia para endenter a síndrome de Crouzon. Associação craniofacial infantil.
- NIH (2016). Síndrome de Crouzon. Obtido em Genetics Home Reference.
- Orphanet (2013). Doença de Crouzon. Obtido da Orphanet.
- Hospital Infantil de Seattle. (2016). Sintomas da síndrome de Crouzon. Obtido no Hospital Infantil de Seattle.