A succinato desidrogenase é uma enzima essencial para o metabolismo celular, atuando na cadeia de transporte de elétrons e na produção de energia na forma de ATP. Sua estrutura é composta por quatro subunidades e cofatores como o grupo heme e o FAD. A regulação da atividade da succinato desidrogenase é crucial para o equilíbrio energético da célula e é afetada por diversos fatores, como a disponibilidade de substratos e a presença de inibidores. Alterações na função desta enzima estão associadas a doenças hereditárias, como a síndrome de Cowden e a paraganglioma, além de ser um marcador de prognóstico em diversos tipos de câncer. Portanto, o estudo da succinato desidrogenase é fundamental para compreender a fisiologia celular e o desenvolvimento de novas terapias para doenças relacionadas ao metabolismo energético.
Importância da succinato desidrogenase no metabolismo energético e no ciclo de Krebs.
A succinato desidrogenase, também conhecida como complexo II, desempenha um papel fundamental no metabolismo energético das células e no ciclo de Krebs. Este complexo enzimático está localizado na membrana interna das mitocôndrias e participa da cadeia de transporte de elétrons, que é responsável pela produção de ATP, a principal fonte de energia das células.
Uma das principais funções da succinato desidrogenase é catalisar a oxidação do succinato a fumarato no ciclo de Krebs. Este processo gera moléculas de FADH2, que são posteriormente utilizadas na cadeia de transporte de elétrons para a produção de ATP. Assim, a succinato desidrogenase é essencial para a geração de energia celular.
Além disso, a succinato desidrogenase também desempenha um papel crucial na regulação do metabolismo celular. Esta enzima está envolvida na regulação da síntese de ácidos graxos e na resposta ao estresse oxidativo, ajudando a manter o equilíbrio energético das células.
Em relação às doenças, mutações na succinato desidrogenase estão associadas a distúrbios metabólicos, como a deficiência de succinato desidrogenase e a síndrome de Cowden, que podem causar problemas no metabolismo energético e no ciclo de Krebs.
Em suma, a succinato desidrogenase é uma enzima essencial no metabolismo energético das células, desempenhando um papel crucial na produção de ATP e na regulação do metabolismo celular. Sua importância no ciclo de Krebs e na cadeia de transporte de elétrons a torna fundamental para a manutenção da saúde e do funcionamento adequado das células.
Enzimas envolvidas na produção de succinato e Malato são respectivamente identificadas.
As enzimas envolvidas na produção de succinato e malato são conhecidas como succinato desidrogenase e malato desidrogenase, respectivamente. A succinato desidrogenase é uma enzima localizada na membrana interna das mitocôndrias e desempenha um papel crucial na cadeia de transporte de elétrons durante a respiração celular. Ela catalisa a reação de conversão do succinato em fumarato, enquanto simultaneamente reduz o coenzima FAD em FADH2.
A malato desidrogenase, por sua vez, atua na conversão do malato em oxaloacetato, participando do ciclo do ácido cítrico. Essa enzima é encontrada no citosol e nas mitocôndrias e desempenha um papel importante na produção de energia através da oxidação dos substratos.
Essas enzimas desempenham papéis essenciais no metabolismo celular, garantindo a produção de intermediários importantes como o succinato e o malato. Alterações na atividade ou expressão dessas enzimas podem levar a distúrbios metabólicos e doenças relacionadas ao metabolismo energético.
Reação catalisada pela succinato desidrogenase: qual é o seu papel no metabolismo celular?
A succinato desidrogenase é uma enzima chave no metabolismo celular, desempenhando um papel fundamental na cadeia de transporte de elétrons e na produção de energia nas células. Essa enzima está envolvida na oxidação do succinato para fumarato, um processo crucial na respiração celular.
A reação catalisada pela succinato desidrogenase envolve a transferência de elétrons do succinato para o FAD, resultando na formação de fumarato e FADH2. Esses elétrons são então transferidos para a cadeia de transporte de elétrons, onde são utilizados para gerar ATP, a principal fonte de energia celular.
Além de sua função na produção de energia, a succinato desidrogenase também desempenha um papel importante na regulação do metabolismo celular. Ela atua como um regulador chave do ciclo do ácido cítrico, controlando a disponibilidade de intermediários metabólicos e garantindo a homeostase celular.
Alterações na atividade da succinato desidrogenase estão associadas a várias doenças, incluindo deficiências metabólicas hereditárias e câncer. Mutações nessa enzima podem levar a distúrbios no metabolismo celular e ao desenvolvimento de tumores, tornando-a um alvo potencial para terapias direcionadas.
Sua disfunção está associada a várias doenças, destacando a importância dessa enzima no funcionamento adequado das células.
Principais características das enzimas: quais são as 4 propriedades fundamentais?
As enzimas são proteínas que atuam como catalisadores em reações químicas dentro das células. Elas possuem quatro propriedades fundamentais que as distinguem de outras moléculas: especificidade, eficiência, regulação e capacidade de serem reutilizadas.
A especificidade das enzimas refere-se à capacidade de cada enzima de catalisar uma reação química específica, atuando apenas sobre um substrato específico. Isso ocorre devido à forma tridimensional única da enzima, que se encaixa perfeitamente no substrato.
A eficiência das enzimas está relacionada à capacidade de acelerar as reações químicas em até milhões de vezes. Isso ocorre devido à diminuição da energia de ativação necessária para a reação ocorrer, aumentando a velocidade da reação.
A regulação das enzimas é essencial para manter o equilíbrio metabólico dentro das células. As enzimas podem ser ativadas ou inibidas por moléculas reguladoras, garantindo que as reações ocorram no momento e na quantidade adequada.
Por fim, a capacidade de as enzimas serem reutilizadas é uma característica importante para a economia de recursos dentro das células. Após catalisar uma reação, a enzima permanece intacta e pronta para catalisar novas reações, sendo utilizada várias vezes.
Succinato desidrogenase: estrutura, função, regulação, doenças
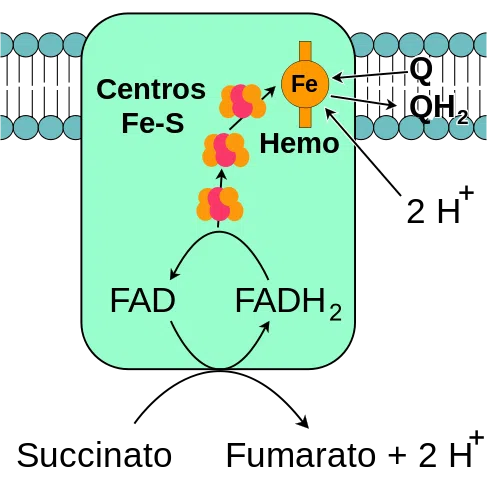
A Succinato desidrogenase (SDH) é uma enzima localizada na membrana interna da mitocôndria, essencial para o ciclo do ácido cítrico e a cadeia respiratória. Sua estrutura é composta por quatro subunidades (A, B, C e D) e possui como cofatores o FAD e o ferro enxofre.
A principal função da SDH é catalisar a oxidação do succinato a fumarato, produzindo FADH2, que será utilizado na cadeia respiratória para a produção de ATP. Além disso, a SDH está envolvida na regulação do metabolismo celular, atuando como um sensor de oxigênio.
Alterações na atividade da Succinato desidrogenase estão relacionadas a diversas doenças, como a síndrome de Cowden, o glioma paraganglioma e o carcinoma renal hereditário. Essas doenças estão associadas a mutações nos genes que codificam as subunidades da enzima, afetando sua função e levando a distúrbios no metabolismo celular.
Succinato desidrogenase: estrutura, função, regulação, doenças
A succinato desidrogenase ( SDH ), também conhecida como complexo II da cadeia de transporte de elétrons, é um complexo proteico mitocondrial com atividade enzimática que funciona tanto no ciclo de Krebs quanto na cadeia de transporte de elétrons (respiração celular).
É uma enzima que está presente em todas as células aeróbicas. Nos eucariotos, é um complexo intimamente associado à membrana mitocondrial interna, enquanto nos procariontes é encontrado na membrana plasmática.
O complexo succinato desidrogenase, descoberto por volta de 1910 e purificado pela primeira vez em 1954 por Singer e Kearney, foi extensivamente estudado por várias razões:
– funciona tanto no ciclo de Krebs (ciclo do ácido cítrico ou do ácido tricarboxílico) quanto na cadeia de transporte de elétrons (catalisa a oxidação do succinato em fumarato)
– sua atividade é regulada por diferentes ativadores e inibidores e
– é um complexo associado a: ferro não ligado a um grupo heme, enxofre lábil e dinucleotídeos de flavina adenina (FAD)
É codificado pelo genoma nuclear e ficou provado que mutações nos quatro genes que codificam cada uma de suas subunidades (A, B, C e D) resultam em vários quadros clínicos, ou seja, podem ser bastante negativos do ponto de vista da integridade física dos seres humanos.
Estrutura
O complexo enzimático succinato desidrogenase consiste em quatro subunidades (heterotetrâmero) codificadas pelo genoma nuclear, tornando-o o único complexo de fosforilação oxidativa na cadeia de transporte de elétrons que não possui nenhuma subunidade codificada pelo genoma mitocondrial.
Além disso, esse complexo é o único que não bombeia prótons através da membrana mitocondrial interna durante sua ação catalítica.
De acordo com estudos realizados com base no complexo enzimático de células cardíacas porcinas, o complexo succinato desidrogenase consiste em:
– um hidrófilo ” cabeça ” , que se estende a partir da membrana mitocondrial interna na matriz mitocondrial e
– um hidrofóbico ” cauda ” que é incorporado na membrana mitocondrial interna e que tem um pequeno segmento que se projecta para o espaço intermembranar das mitocôndrias solúvel
Estrutura da porção hidrofílica
A cabeça hidrofílica é composta pelas subunidades SdhA (70 kDa) e SdhB (27 kDa) (Sdh1 e Sdh2 em levedura) e compreende o centro catalítico do complexo.
As subunidades SdhA e SdhB contêm os cofatores redox que participam da transferência de elétrons para a ubiquinona (coenzima Q10, uma molécula que transporta elétrons entre os complexos respiratórios I, II e III).
A subunidade SdhA possui um cofator FAD (uma coenzima que participa de reações de redução de óxido) covalentemente ligado à sua estrutura, exatamente no local de ligação do succinato (principal substrato da enzima).
A subunidade SdhB possui 3 centros de ferro-enxofre (Fe-S) que mediam a transferência de elétrons para a ubiquinona. Um dos centros, 2Fe-2S, fica próximo ao local FAD da subunidade SdhA e os outros (4Fe-4S e 3Fe-4S) são adjacentes ao primeiro.
Deve-se notar que estudos estruturais indicam que a subunidade SdhB forma a interface entre o domínio catalítico hidrofílico e o domínio “âncora” da membrana (hidrofóbico) do complexo.
Estrutura da porção hidrofóbica
O domínio da membrana do complexo, como declarado, consiste nas subunidades SdhC (15 kDa) e SdhD (12-13 kDa) (Sdh3 e Sdh4 em levedura), que são proteínas integrais da membrana, cada uma formada por 3 hélices transmembranares .
Esse domínio contém uma parte do heme b ligada à interface entre as subunidades SdhC e SdhD, onde cada uma fornece um dos dois ligantes de histidina que os mantêm unidos.
Dois locais de ligação à ubiquinona foram detectados nesta enzima: um com alta afinidade e outro com baixa afinidade.
O local de alta afinidade, conhecido como Qp (p para proximal ), enfrenta a matriz mitocondrial e é composto de resíduos de aminoácidos específicos localizados nas subunidades SdhB, SdhC e SdhD.
O local de baixa afinidade, também chamado de Qd (d para distal ), está na porção da membrana mitocondrial interna onde o complexo é inserido, mais próximo do espaço intermembranar, ou seja, mais distante da matriz organelar.
Tomado em conjunto, o complexo total tem um peso molecular próximo a 200 kDa e foi determinado que possui uma proporção de 4,2-5,0 nanomoles de flavina para cada miligrama de proteína e 2-4 g de ferro para cada mole de flavina.
Função
O complexo enzimático succinato desidrogenase cumpre uma função importante nas mitocôndrias, pois participa não apenas do ciclo de Krebs (onde participa da degradação do acetil-CoA), mas também faz parte da cadeia respiratória, essencial para a produção de energia sob a forma de ATP.
Em outras palavras, é uma enzima essencial para o metabolismo intermediário e a produção aeróbica de ATP.
– É responsável pela oxidação do succinato em fumarato no ciclo do ácido cítrico
– Alimenta o complexo III da cadeia de transporte de elétrons com os elétrons derivados da oxidação do succinato, o que ajuda a reduzir o oxigênio e formar a água
– O transporte de elétrons gera um gradiente eletroquímico através da membrana mitocondrial interna, o que favorece a síntese de ATP
Como alternativa, os elétrons podem ser usados para reduzir moléculas de um pool de ubiquinonas, produzindo os equivalentes redutores necessários para reduzir os ânions superóxido, originários da mesma cadeia respiratória ou provenientes de fontes exógenas.
Como funciona?
A subunidade A do complexo (aquela que está covalentemente ligada à coenzima FAD) se liga aos substratos fumarato e succinato, bem como seus reguladores fisiológicos, oxalacetato (inibidor competitivo) e ATP.
O ATP desloca a ligação entre o oxaloacetato e o complexo SDH, e então os elétrons que são “passados” do succinato para a subunidade SdhA são transferidos para os grupos de átomos de ferro e enxofre presentes na subunidade SdhB por meio da coenzima FAD.
A partir da subunidade B, esses elétrons atingem os locais heme b das subunidades SdhC e SdhD, de onde são “entregues” às coenzimas da quinona através de seus locais de ligação à quinona.
O fluxo eletrônico do succinato através desses transportadores para o aceitador final, que é oxigênio, é acoplado à síntese de 1,5 moléculas de ATP para cada par de elétrons através da fosforilação ligada à cadeia respiratória.
Defeitos na enzima
Foi relatado que mutações no gene que codifica a subunidade A do complexo succinato desidrogenase causam encefalopatias durante a infância, enquanto mutações nos genes que codificam as subunidades B, C e D têm sido associadas à formação de tumores.
Regulamento
A atividade do complexo succinato desidrogenase pode ser regulada por modificações pós-traducionais, como fosforilação e acetilação , embora também possa ocorrer inibição do sítio ativo.
A acetilação de alguns resíduos de lisina pode diminuir a atividade dessa enzima e esse processo é realizado por uma enzima desacetilase conhecida como SIRT3; a fosforilação tem o mesmo efeito na enzima.
Além dessas modificações, o complexo SDH também é regulado pelos intermediários do ciclo de Krebs, especificamente oxalacetato e succinato . O oxalacetato é um poderoso inibidor, enquanto o succinato favorece a dissociação do oxalacetato, funcionando como ativador.
Deficiência de succinato desidrogenase
A deficiência de succinato desidrogenase é uma anormalidade ou distúrbio da cadeia respiratória mitocondrial. Essa deficiência é causada por mutações nos genes SDHA (ou SDHAF1), SDHB, SDHC e SDHD.
Diferentes investigações mostraram mutações homozigotas e heterozigotas nesses genes, especialmente SDHA. Mutações desses genes causam substituições de aminoácidos na proteína (em qualquer uma das subunidades SDHA, B, C ou D) ou, alternativamente, codificam proteínas anormalmente curtas.
Consequentemente, substituições anormalmente curtas de aminoácidos e codificações de proteínas levam a distúrbios ou alterações da enzima SDH, causando uma falha na capacidade ideal das mitocôndrias de produzir energia. Isso é o que os cientistas chamam de distúrbio da cadeia respiratória mitocondrial.
Este distúrbio pode ser expresso fenotipicamente em humanos de várias maneiras. Os mais conhecidos são: deficiência ou falta de desenvolvimento lingüístico, quadriplegia espástica, contrações musculares involuntárias (distonia), fraqueza muscular e cardiomiopatias, entre outros problemas relacionados.
Alguns pacientes com deficiência de succinato desidrogenase podem desenvolver a doença de Leigh ou a síndrome de Kearns-saire.
Como é detectada a deficiência de succinato desidratado?
Alguns estudos sugerem o uso de análises e testes histoquímicos qualitativos, bem como análises quantitativas, enzimáticas e bioquímicas da cadeia respiratória. Outros, por sua vez, sugerem a amplificação completa por meio da reação em cadeia da polimerase (PCR) dos exons das subunidades em estudo e, em seguida, o respectivo sequenciamento.
Doenças relacionadas
Há um grande número de expressões fenotípicas produzidas por distúrbios da cadeia respiratória mitocondrial, devido à deficiência de succinato desidrogenase. No entanto, quando se trata de síndromes ou doenças, são mencionados os seguintes.
Síndrome de Leigh
É uma doença neurológica progressiva, associada a mutações no genoma nuclear (neste caso, succinato desidrogenase), que afetam o complexo piruvato desidrogenase até o caminho da fosforilação oxidativa.
Os sintomas aparecem antes do primeiro ano de idade do indivíduo, mas em casos atípicos, os primeiros sintomas foram observados durante a adolescência.
Entre os sintomas mais comumente observados estão: hipotonia com perda de controle da cabeça, movimentos involuntários, vômitos recorrentes, problemas respiratórios, incapacidade de mover o globo ocular, sinais piramidais e extrapiramidais, entre outros. Convulsões não são muito comuns.
É possível que a doença possa ser detectada nos diagnósticos pré-natais. Não há cura ou tratamento específico conhecido, mas alguns especialistas sugerem tratamentos com certas vitaminas ou cofatores.
Tumor estromal gastrointestinal (GIST)
Comumente chamado GIST, é um tipo de tumor do trato gastrointestinal, que geralmente se desenvolve em áreas como o estômago ou intestino delgado. Acredita-se que a causa disso se deva a um determinado grupo de células altamente especializadas chamadas células ICC ou células intersticiais Cajal.
Outras considerações sobre a causa dos GISTs são as mutações de certos tipos de genes, que segundo alguns autores causam 90% dos tumores. Os genes envolvidos são: genes KIT, PDGFRA, succinato desidrogenase (SDH) – deficientes.
A deficiência de succinato desidrogenase (SDH) ocorre principalmente em mulheres jovens, produz tumores no estômago e, com relativa frequência, metástase nos gânglios linfáticos. Uma pequena porcentagem ocorre em crianças e, na maioria dos casos, é devido à falta de expressão da subunidade SDHB.
Síndrome de Kearns-Sayre
Foi determinado que alguns pacientes com deficiências de succinato desidrogenase podem manifestar a síndrome de Kearns-Sayre. Esta doença está relacionada a distúrbios mitocondriais e é caracterizada pela ausência de movimento dos globos oculares.
Outras características desta doença são retinite pigmentosa, surdez, cardiomiopatia e distúrbios do sistema nervoso central. Esses sintomas geralmente são observados antes do paciente completar 20 anos. Não há diagnóstico pré-natal conhecido para essa condição.
Também não há cura conhecida para esta doença. O tratamento é paliativo, ou seja, funciona apenas para reduzir os efeitos da doença, não para curá-la. Por outro lado, embora dependa do número de órgãos afetados e da atenção médica recebida, a expectativa de vida é relativamente normal.
Referências
- Ackrell, BA, Kearney, EB e Singer, TP (1978). [47] Succinato desidrogenase de mamíferos. In Methods in enzimology (Vol. 53, pp. 466-483). Academic Press.
- Brière, JJ, Favier, J., Ghouzzi, VE, Djouadi, F., Benit, P., Gimenez, AP e Rustin, P. (2005). Deficiência de succinato desidrogenase em humanos. Cellular and Molecular Life Sciences CMLS, 62 (19-20), 2317-2324.
- Cecchini, G., Schröder, I., Gunsalus, RP, & Maklashina, E. (2002). Succinato desidrogenase e fumarato redutase de Escherichia coli. Biochimica et Biophysica Acta (BBA) -Bioenergetics, 1553 (1-2), 140-157.
- Hatefi, Y. & Davis, KA (1971). Succinate desidrogenase. I. Purificação, propriedades moleculares e subestrutura. Bioquímica, 10 (13), 2509-2516.
- Hederstedt, LARS e Rutberg, LARS (1981). Succinato desidrogenase – uma revisão comparativa. Revisões microbiológicas, 45 (4), 542.
- Nelson, DL, Lehninger, AL, & Cox, MM (2008). Princípios de Lehninger da bioquímica. Macmillan.
- Rutter, J., Winge, DR, & Schiffman, JD (2010). Succinate desidrogenase – montagem, regulação e papel em doenças humanas. Mitocôndria, 10 (4), 393-401.